


Bei Dysmorphie-Retardierungs-Syndromen fällt es oft schwer, eine klinische Diangose zu formulieren. Ein Fallbeispiel eines 22-jährigen Patienten.
Anamnese und Befund
Dieser 1989 geborene Junge wird seit dem 1. Lebensjahr wegen eines unklaren Dysmorphie-Retardierungs-Syndroms in einer neuropädiatrischen Schwerpunkt-Praxis betreut. Er ist das einzige Kind gesunder Eltern, einer thailändischen Mutter und eines deutschen Vaters. Ähnliche Entwicklungsstörungen sind in der Familie nicht bekannt. Nach unauffälliger Schwangerschaft erfolgte die Geburt komplikationslos per Sectio am Termin. In den ersten Lebensmonaten fielen eine opisthotone Haltung und Fausten auf. Nach einem Sturz mit Schädelfraktur im Alter von 4 Monaten wurden eine statomotorische Retardierung, muskuläre Hypotonie und Schulterretraktion diagnostiziert. Mit 9 Monaten traten Schreckhaftigkeit und athetoide Bewegungsmuster auf. In seiner motorischen Entwicklung erreichte er das Drehen, mit 20 Monaten sprach er wenige einzelne Worte.
Im weiteren Verlauf kam es zu motorischen und sprachlichen Rückschritten. Seit dem 3. Lebensjahr trat eine Epilepsie mit zunächst fiebergebundenen, dann auch afebrilen generalisierten tonisch-klonischen Anfällen, später auch myoklonischen Anfällen auf, die sich als therapieresistent erwies. Die Versorgung mit einer perkutanen Gastroenterostomie sowie mit einem Tracheostoma wurde erforderlich.
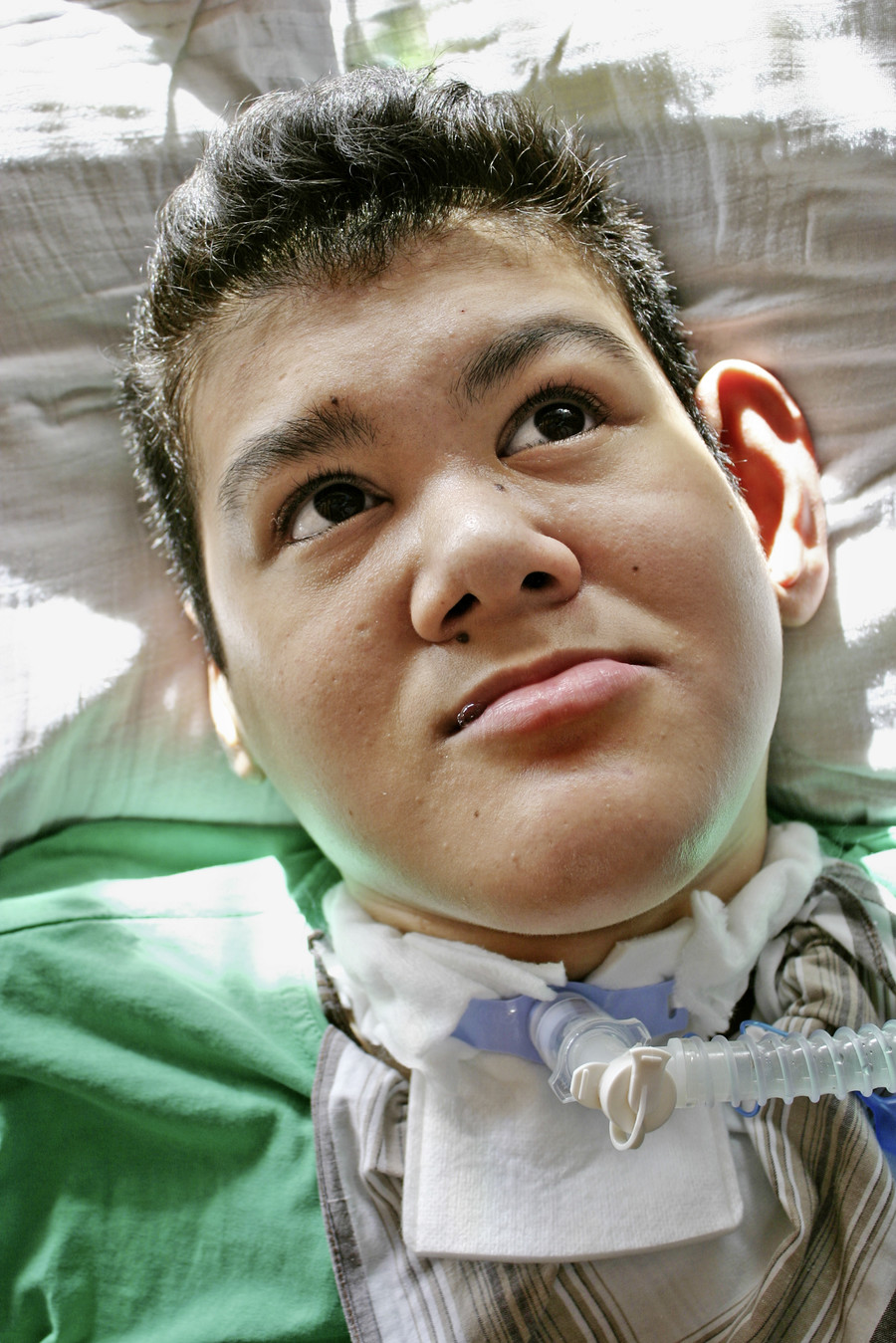
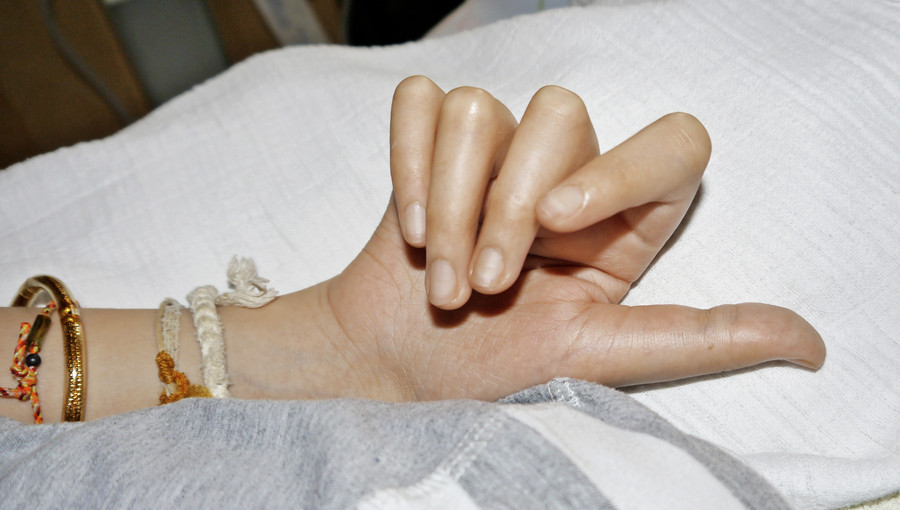
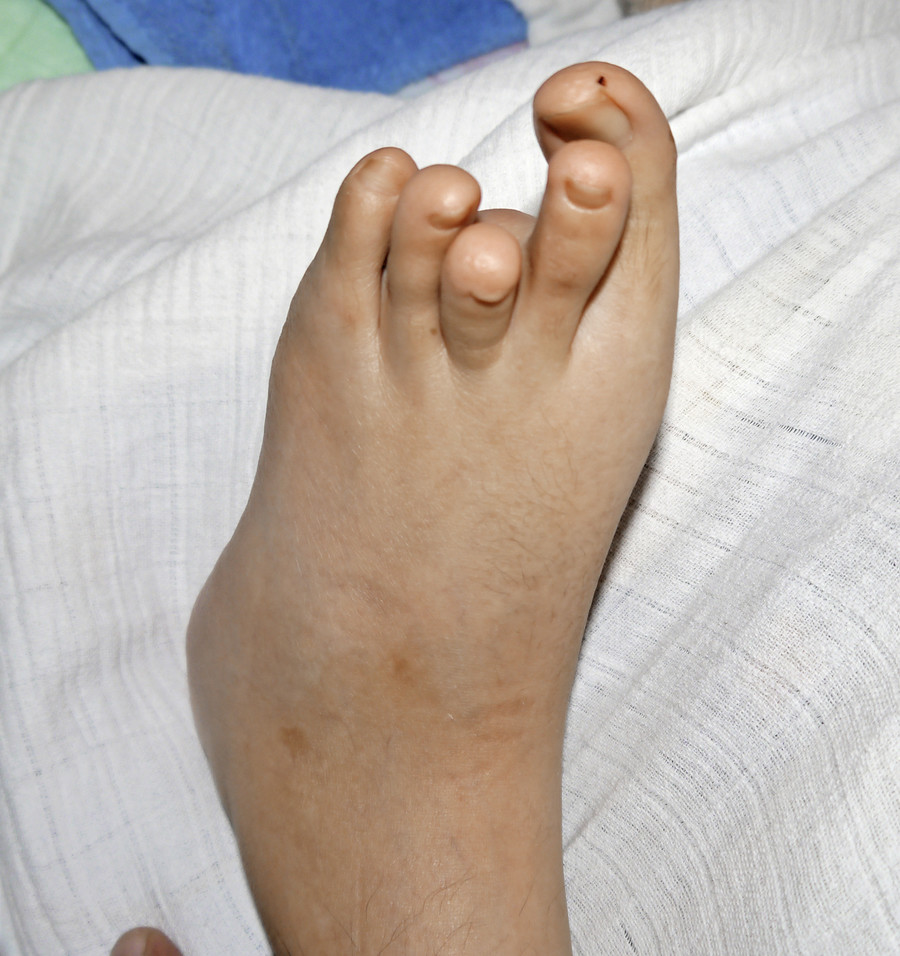
Bei der körperlichen Untersuchung im Alter von 22 Jahren weist der schwer mehrfachbehinderte Patient neben einer spastischen Tetraparese eine ausgeprägte Gesichtsasymmetrie, asymmetrische dysplastische Ohren, eine kleine Oberlippe und prominente Unterlippe, einen hohen Gaumen, eine erhebliche Skoliose sowie lange Finger und lange Großzehen auf (Abb. 1, 2).
Craniale MR-Tomographien zeigten zunächst keine wegweisenden Besonderheiten. Bei einer Kontrolle mit 21 Jahren werden eine Kleinhirnatrophie, eine flaue Signalgebung im zentralen Hirnstamm und erweiterte perivaskuläre Räume beschrieben.
Molekulargenetisch fanden sich bei Analyse der mtDNA keine Hinweise auf eine Myoklonusepilepsie mit ragged red fibers (MERRF), auch waren weder eine Mutation des SCN1A-Gens (Dravet-Syndrom) noch des SCL2A1-Gens (Glukosetransporter-Defekt) nachweisbar. Der Methylierungstest sprach gegen das Vorliegen eines Angelman-Syndroms, ebenso fand sich im Array-CGH keine relevante chromosomale Mikroaberration. So blieb die Ätiologie der schweren Retardierung und der Epilepsie über 20 Jahre lang unklar.
Bei der differenzialdiagnostischen Abklärung eines Dysmorphie-Retardierungs-Syndroms wird der untersuchende Arzt stets zunächst bemüht sein, nach Anamnese und Befund eine klinische Verdachtsdiagnose zu stellen, die dann durch gezielte Zusatzuntersuchungen zu bestätigen oder weitgehend auszuschließen ist. In vielen Fällen gelingt diese Hypothesenbildung jedoch nicht, da wegweisende klinische Befunde nicht erkennbar sind oder wegen der Seltenheit des Krankheitsbildes vom Untersucher nicht unmittelbar zugeordnet werden können. Die Abklärung derartiger unklarer Fehlbildungssyndrome oder auch einer nicht syndromalen Retardierung durch eine Chromosomenanalyse ist seit einigen Jahren durch die vergleichende genomische Hybridisierung (Array-CGH)erheblich verfeinert worden.
Nachdem die genannten diagnostischen Schritte bei dem hier vorgestellten Patienten keine Klärung der Ursache ergab, wurde eine molekulargenetische Panel-Diagnostik hinsichtlich genetisch bedingter Epilepsien durchgeführt. Bei diesem Verfahren werden mit Hilfe einer Hochdurchsatz-Sequenzierung sämtliche für eine Krankheit in Betracht kommenden Gene gleichzeitig parallel analysiert. Es können somit beispielsweise bei Vorliegen einer genetisch bedingten Epilepsie derzeit etwa 400 Gene parallel analysiert werden. Die Auswertung bezüglich krankheitsverursachender Mutationen erfolgt dabei ausschließlich für mit der klinischen Symptomatik in Zusammenhang gebrachte Gene.
Bei dieser Panel-Diagnostik zeigte sich bei unserem Patienten in Exon 3 des SMS-Gens auf Chromosom Xp22.11 eine hemizygote Missense-Mutation, die auf Proteinebene zum Ersatz des Phenylalanins durch Leucin an Position 58 führt (c.174T>A; p.F58L) [3]. In Abbildung 3 ist die Mutation dargestellt. Das SMS-Gen kodiert für die Sperminsynthase. Dieses Enzym wandelt Spermidin in Spermin um. Spermin und Spermidin interagieren mit Aminosäuren und Proteinen und modulieren sowohl Kaliumkanäle, Kalziumkanäle als auch AMPA-Rezeptoren, zudem haben sie Einfluss auf zerebelläre Regelkreise [1].
Diese Sequenzveränderung war in der Literatur noch nicht beschrieben. Eine Analyse dieser Variante mit dem Vorhersageprogramm "MutationTaster" stuft sie als krankheitsverursachend ein. Die Mutation wird als wahrscheinlich pathogen angesehen, da sie nur 4 Nukleotide von der Akzeptor-Splice-Site des Introns 2 entfernt liegt und somit möglicherweise ein aberrantes Splicing (also das Herausschneiden der Exons und deren Verknüpfung mit der fertigen messenger RNA) verursacht. Des Weiteren befindet sich die Mutation in einem phylogenetisch hochkonservierten Bereich des SMS-Proteins innerhalb der N-terminalen Domäne, die für die Bildung eines Homodimers und für die enzymatische Aktivität des Proteins essentiell ist. Die durchgeführte Segregationsanalyse ergab zudem, dass die Mutter nicht Trägerin dieser Mutation ist und es sich somit um eine Neumutation ("de novo"-Mutation) handelt.
In der Literatur sind derzeit 11 Patienten mit Mutationen im SMS-Gen beschrieben. Für manche dieser Mutationen wurde ein Aktivitätsverlust der Sperminsynthase nachgewiesen [1, 2]. Die entsprechende biochemische Diagnostik bei unserem Patienten im Labor des Greenwood Genetic Center, South Carolina (C. Schwartz, PhD) ergab einen erniedrigten Spermin/Spermidin-Quotienten von 0,37 (Durchschnitt in 3 Kontrollseren: 2,0) und eine extrem niedrige Sperminsynthaseaktivität.
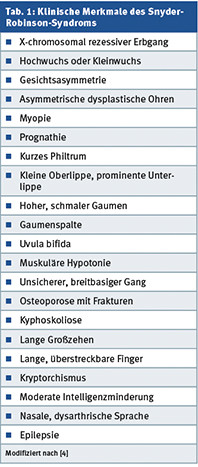
Somit führen molekulargenetische und biochemische Diagnostik übereinstimmend zu der Diagnose eines Snyder-Robinson-Syndroms (OMIM #309583), einer besonderen, syndromalen Form der X-chromosomalen Intelligenzminderung. Dies wissend sieht der Untersucher bei klinischer Re-Evaluation des Patienten nun tatsächlich zahlreiche klinische Symptome, die typisch für dieses Krankheitsbild sind, die aber von den im Laufe der Jahre zahlreich konsultierten Ärzten wegen der Seltenheit des Snyder-Robinson-Syndroms nicht als diagnostisch wegweisend realisiert wurden (Tab. 1). Allerdings ist der hier vorgestellte Patient schwerer betroffen als die bisher beschriebenen [4].
Der Einsatz der neuen molekulargenetischen Untersuchungsmethode, des sog. next generation sequencing im Rahmen einer Panel-Diagnostik, hat also bei diesem Patienten nach vielen Jahren der Ursachensuche zur Diagnose geführt. Eine Diagnosestellung zu einem früheren Zeitpunkt hätte eine genetische Beratung der Eltern über das relativ geringe Wiederholungsrisiko von unter 5 % (bei möglichem Keimbahnmosaik [5]) und das Angebot einer gezielten pränatalen Diagnostik für zukünftige Schwangerschaften ermöglicht.
Trotz des späten Diagnosezeitpunkts ergaben sich für den Patienten therapeutische Konsequenzen. Da bekannt ist, dass AMPA-Rezeptoren in der Pathophysiologie des Snyder-Robinson-Syndroms eine Rolle spielen, wurde (nach Zulassung in Deutschland) eine Therapie mit Perampanel begonnen. Perampanel gehört zu den AMPA-Rezeptor-Antagonisten, welche der durch Glutamat vermittelten Hyperexzitabilität entgegenwirken. Diese Medikation zeigte eine deutlich bessere Wirksamkeit als die bis dahin eingesetzten Antiepileptika.
- Bei Dysmorphie-Retardierungs-Syndromen gelingt es auch dem erfahrenen Kliniker oft nicht, anhand von Anamnese, körperlichem Befund und apparativen Zusatzuntersuchungen eine klinische Diagnose zu formulieren.
- Die Chromosomenanalyse als die in dieser Situation diagnostisch ertragreichste humangenetische Screening-Untersuchung wurde in den letzten Jahren durch die vergleichende genomische Hybridisierung (Array-CGH) abgelöst.
- Auch die Array-CGH liefert nur in ca. 15 % der untersuchten Patienten eine Klärung der Ursache.
- Neuere humangenetische Methoden wie z. B. die sogenannte Panel-Diagnostik ermöglichen in vielen Fällen eine Ursachenklärung auch für sehr seltene genetische Erkrankungen – gelegentlich mit überraschenden therapeutischen Konsequenzen für den betroffenen Patienten, nahezu immer mit Konsequenzen für die genetische Beratung der betroffenen Familie.
Isabelle Steiner¹, Rüdiger Lorenz² | ¹Center for Genomics and Transcriptomics (CeGaT), Tübingen; ²Bad Wildungen

Erschienen in: Kinderärztliche Praxis, 2013; 84 (6) Seite 374-378


