
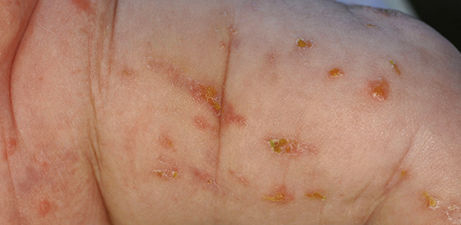

Streifenförmig angeordnete Papulovesikel und Erosionen an der Haut eines neugeborenen Mädchens. Wie lautet Ihre Diagnose? Ein Fallbeispiel aus der Rubrik "Der diagnostische Blick".
Anamnese und Befund
Ein 8 Wochen alter weiblicher Säugling wird mit seit 2 Tagen nach der Geburt bestehenden, vorwiegend linear angeordneten, gelblich-krustös belegten Papeln und Erosionen sowie erythematösen Blasen an den Extremitäten bei sonst völliger Gesundheit vorgestellt.
Die nicht miteinander verwandten Eltern des Mädchens berichten über eine komplikationslose Schwangerschaft und termingerechte Spontangeburt sowie einen guten Entwicklungszustand des Kindes. Die postnatal bestimmten Routinelaborparameter, Differentialblutbild und die mikrobiologische Untersuchung des Bläscheninhaltes waren unauffällig.
Wie die Mutter berichtet, waren auch bei ihr selbst wenige Tage nach ihrer Geburt solche Hautveränderungen aufgetreten, die dann im Alter von etwa 5 Jahren abheilten. Als Restveränderung bestehen jetzt noch kaum sichtbare, leicht atrophe Hypopigmentierungen am Unterschenkel. Niemand sonst in der Familie ist von ähnlichen Hautveränderungen betroffen.
Bei der Untersuchung des 8 Wochen alten Mädchens zeigen sich an allen Extremitäten in überwiegend striärer Anordnung entlang der Blaschko-Linien gelblich-krustös belegte Erosionen und Papulovesikel auf gerötetem Grund sowie bräunlich-rötliche makulöse Hautveränderungen mit zum Teil collerette-artiger Schuppenkrause (in Abheilung befindliche Läsionen) (Abb.1). Am vorderen oberen Rumpf sieht man ein eher ekzematöses Bild mit teilweise follikulär gebundenen Papeln sowie feinlamellärer Schuppung. Das übrige Integument inklusive Palmae, Plantae und einsehbarer Schleimhäute ist erscheinungsfrei.
Incontinentia pigmenti (IP, Bloch-Sulzberger-Syndrom) ist eine seltene X-chromosomal-dominant vererbbare neuroektodermale Genodermatose mit einer Inzidenz von 1/40.000 Einwohner/Jahr. Sie tritt fast ausschließlich bei weiblichen Individuen auf, betroffene männliche Feten sterben meist intrauterin ab. Die Erkrankung ist charakterisiert durch typische, bereits in den ersten Lebenstagen bis -wochen auftretende Hautveränderungen entlang der Blaschko-Linien, die einen stadienhaften, teils überlappenden Verlauf aufweisen.
- Stadium 1: Linear angeordnete Vesikel und Bullae auf erythematösem Grund
- Stadium 2: Hyperkeratotische, verruköse, im Verauf abflachende Plaques, akral beginnend
- Stadium 3: spritzerartige, teils retikuläre Hyperpigmentierungen
- Stadium 4 (6. LJ bis Erwachsenenalter): Hypopigmentierte, atrophe Areale, insbesondere an den unteren Extremitäten
Zusätzlich können weitere Organsysteme wie Haare, Nägel, Zähne, ZNS, Augen und Skelett betroffen sein [1, 4, 6].
Genetische Diagnostik
Ursächlich liegen der IP Mutationen im IKBKG (inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase gamma)-Gen (Xq28), alternativ NEMO (NF-kappa-B essential modulator)-Gen genannt, zugrunde. Dieses Gen kodiert für ein Protein, das sowohl essentiell für den Schutz vor TNF-alpha-induzierter Apotose als auch verantwortlich für die Regulation verschiedener Zytokine, Chemokine und Adhäsionsmoleküle ist. Molekulargenetische Analysen des IKBKG-Gens zeigen dabei in ca. 80 % der Fälle eine intragenetische Deletion von Exon 4 bis 10. Die Mutationen sind nur mit dem Leben vereinbar, wenn sie als Mosaik vorliegen wie z. B. bei Frauen im Rahmen der X-chromosomalen Inaktivierung (Lyon-Hypothese) während der Embryonalentwicklung. Mosaike können bei männlichen Individuen im Rahmen des Klinefelter-Syndroms (47,XXY Karyotyp), durch Chromatiden-Mutation oder durch frühe somatische Mutation vorkommen [1, 3, 4, 6].
In dem geschilderten Fall konnten wir anhand des charakteristischen klinischen Untersuchungsbefundes bei dem Kind, der positiven Familienanamnese bei der Mutter sowie durch den molekulargenetischen Nachweis der typischen Deletion von Exon 4 bis 10 im IKBKG-Gen bei dem Säugling die Diagnose Incontinentia pigmenti stellen.
Therapie und Verlauf
Unter zunächst antiseptischer Infektionsprophylaxe der Bläschen und Erosionen mit Octenidin Lösung und im weiteren antiseptisch–antiinflammatorischer Lokaltherapie mit Prednicarbat im Wechsel mit Fusidinsäure sowie indifferenter Pflege mit Ungtum leniens DAB 10 kam es zur fast vollständigen Abheilung der Effluoreszenzen.
Zum Zeitpunkt der Vorstellung in unserer Klinik lagen keine weiteren organischen Manifestationen bei dem Mädchen vor. Die Prognose der IP jedoch ist entscheidend von dem Ausmaß der systemischen Beteiligung abhängig, die von sehr diskret ausgeprägten Hautveränderungen bis hin zum Vollbild mit schweren Entwicklungsstörungen variieren kann. Hierüber kann aufgrund des jungen Alters der Patientin noch keine abschließende Aussage getroffen werden. Da ca. 80 % der Patienten assoziierte systemische Manifestationen aufweisen, sind regelmäßige dermatologische, pädiatrisch-neurologische und ophthalmologische Kontrolluntersuchungen sowie eine humangenetische Beratung zu empfehlen [4, 6].
Diskussion
Interessanterweise war die Mutation bei der Mutter trotz sehr suggestiver Anamnese für das Vorliegen einer IP und auch ärztlicher Dokumentation des damaligen klinischen Untersuchungsbefundes nach zweimaliger Wiederholung der molekular-genetischen Untersuchung im Blut nicht detektierbar. Auch klinisch wies die Mutter seit Abheilung der Hautveränderungen (jetzt nur noch kaum sichtbare, leicht atrophe Hypopigmentierungen am Unterschenkel) keine weitere Organbeteiligung auf.
Möglicherweise ist dies durch das Vorliegen eines funktionellen X-chromosomalen Mosaiks im Rahmen der Inaktivierung jeweils eines X-Chromosoms (Lyon-Hypothese) während der Embryonalphase erklärbar, so dass die Mutation z. B. nur noch in den betreffenden residualen Hautläsionen am Unterschenkel nachweisbar ist. Zellen mit einem mutierten aktiven X-Chromosom können außerdem sukzessive durch Apoptose eliminiert werden, was unter anderem den stadienhaften Verlauf mit der Rückbildung der Hautveränderungen bewirkt [2,3].
Des Weiteren könnte der Grad der Ungleichverteilung der X-Chromosom-Inaktivierung ("skewed X-inactivation") eine Rolle spielen, der sich im Laufe des Lebens ändern kann [7]. So findet man bei erwachsenen Frauen erheblich häufiger eine Ungleichverteilung der X-Inaktivierung als bei neugeborenen Mädchen. Liegt die genetische Veränderung bei einem X-chromosomal dominanten Erbgang auf dem genetisch inaktiven Chromosom, kann die Frau der Manifestation der Erkrankung entgehen, die Anlage aber an die Kinder weitergeben [7].
Außerdem kann die typische Deletion von Exon 4 bis 10 im IKBKG-Gen nur in ca. 80 % der Fälle nachgewiesen werden. Bei den übrigen 20 % bestehen möglicherweise sogenannte versteckte Mutationen ("hidden mutations") durch die zweite Kopie des IKBKG-Gens oder die Anwesenheit von hoch homologen IKBKG-Pseudogenen [2].
Um die Diagnose bei der Mutter zu sichern, könnte eine Hautbiopsie aus den atrophen, hypopigmentierten Arealen vom Unterschenkel hilfreich sein, um histologische Untersuchungen vorzunehmen und um die Mutation in Hautzellen (Fibroblasten) zu detektieren [2]. Alternativ kämen auch noch molekulargenetische DNA-Analysen aus anderen ektodermalen Geweben wie Haarwurzeln oder Mundschleimhautzellen in Betracht. Diese Untersuchungen stehen jedoch noch aus.
- Bei in den ersten Lebenstagen bis -wochen auftretenden Hautveränderungen entlang der Blaschko-Linien an Incontinentia pigmenti denken.
- Seltene neuroektodermale Genodermatose mit Mutationen im IKBKG –Gen, die fast ausschließlich weibliche Individuen betrifft; Familienanamese, ggf. molekulargenetische Untersuchung.
- Typischer stadienhafter, teils überlappender Verlauf mit initial linear angeordneten Papulovesikeln und Erosionen auf erythematösem Grund und späterer Veränderung und Abheilung der Hautveränderungen.
- Weitere Organmanifestationen an Haaren, Nägeln, Zähnen, ZNS, Augen und Skelett in 80 % der Fälle.
- Regelmäßige dermatologische, pädiatrisch-neurologische und ophthalmologische Kontrolluntersuchungen sowie eine humangenetische Beratung sind notwendig.
Autoren
Sabine Thoms, Birka Brauns, Michael P. Schön, Martin Mempel | Abteilung Dermatologie, Venerologie und Allergologie, Universitätsmedizin der Georg-August-Universität Göttingen
Erschienen in: Kinderärztliche Praxis, 2011; 82 (6) Seite 377-379


