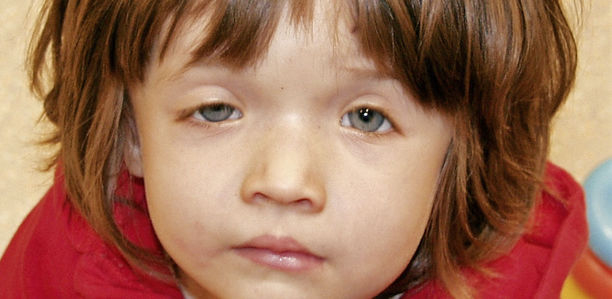
Im Rahme eines Saethre-Chotzen-Syndroms können Kalottendefekte ein wertvolles diagnostisches Kriterium darstellen und der Abgrenzung gegenüber ähnlichen Phänotypen dienen.
Dieser Beitrag steht nur registrierten Benutzern mit Berufsverifizierung zur Verfügung. Jetzt anmelden.Der diagnostische Blick
Ein 20 Monate altes Mädchen mit bilateralen Kalottendefekten